
India has one of the world’s largest consumer markets. With a large and growing population, rising prevalence of chronic diseases, and expanding healthcare infrastructure – including the Ayushman Bharat Digital Mission, the country is becoming an increasingly important market for medical devices.
The growing middle class, continued medical tourism, and increasing adoption of advanced technologies are contributing to the expansion of the Indian market, with the country also serving as a cost-effective location for manufacturing.
India’s regulatory environment for medical devices and in vitro diagnostics (IVDs) has significantly transformed from pharmaceutical-oriented regulations to a specialised, risk-based framework. This evolution prioritises patient safety, product efficacy, and quality standards, positioning India as a key player in the global medical device market.
In part three of our fast-growing markets series, our Head of Regulatory Bruno Gretler shares his insight on India’s regulatory framework, including device classification, market entry approaches, post-market surveillance, and strategies for effective compliance.
An overview of the Indian regulatory framework
The cornerstone of India’s medical device regulatory landscape is the Medical Devices Rules (MDR) 2017 (not to be confused with the EU MDR) – managed by the Central Drugs Standard Control Organisation (CDSCO) under the Ministry of Health and Family Welfare.
MDR 2017 introduced a dedicated, risk-based approach tailored specifically to medical devices, aligning India closely with international standards such as those of the FDA and the European MDR. Compliance involves meeting rigorous standards related to licensing, quality management, clinical evaluation, and post-market surveillance. High-risk devices (Classes C and D) fall under CDSCO’s direct oversight, while lower-risk devices (Classes A and B) are managed by State Licensing Authorities and Notified Bodies.
Further reinforcing this regulatory system, the National Medical Device Policy 2023, emphasises enhancing domestic manufacturing capabilities, fostering innovation, and streamlining market entry without compromising on safety and efficacy.
How are medical devices & IVDs classified in India?
India’s CDSCO provides detailed guidelines covering active, diagnostic, and therapeutic devices, emphasising criteria such as intended use, invasiveness, and delivery methods.
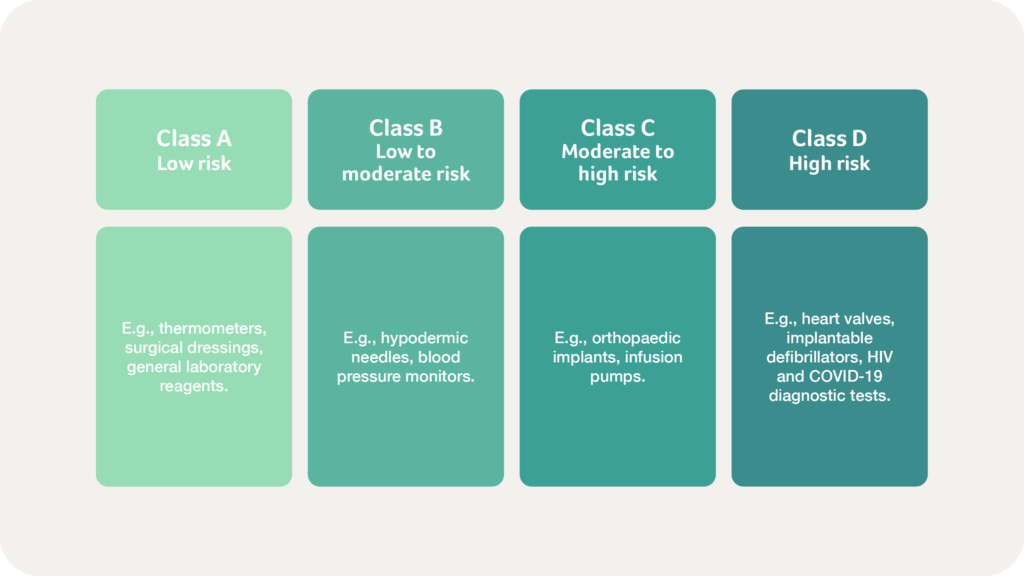
India’s classification system for medical devices and IVDs is based on a risk-oriented approach governed by the Medical Devices Rules (MDR) 2017. This framework aligns broadly with international principles yet is tailored specifically to India’s healthcare needs. Medical devices and IVDs are classified into four categories:
Significant regulatory updates
The introduction of Unique Device Identification (UDI) under MDR 2017 enhances traceability and compliance audits.
Additionally, import licensing requirements are transitioning from voluntary to mandatory, with set deadlines:
- Class A and B devices within 12 months
- Class C and D within 24 months
In 2021, the CDSCO also released specific guidelines for Software as a Medical Device (SaMD), aligned with the International Medical Device Regulators Forum’s (IMDRF) risk-based classification. This classification, which also aligns with the risk classes in the diagram above, determines the regulatory scrutiny required, ensuring appropriate safety and efficacy standards for SaMD products.
CDSCO regularly updates its registry of notified medical devices, essential for ensuring patient safety, promoting innovation, and regulatory compliance, so we recommend that stakeholders consult the CDSCO website directly for the latest updates.
What does the Indian market pathway for medical devices & IVDs involve?
Bringing medical devices or IVDs to market in India requires adherence to a structured regulatory approval process outlined in the Medical Devices Rules (MDR) 2017. The market entry framework balances patient safety and industrial growth, aligns with international best practices, and adapts to local compliance requirements.
The pathway ensures device safety, quality, and efficacy, with steps varying by device classification, origin, and prior global approvals. Manufacturers must stay informed about evolving CDSCO regulations to ensure effective and timely market entry, but generally, the key steps include:
Classifying your device
Manufacturers need to classify their devices according to risk (Class A to D). Lower-risk devices (Class A and B) follow streamlined procedures, whereas higher-risk devices (Class C and D) undergo rigorous evaluations, often involving clinical investigations.
Appointing an Authorised Indian Representative (AIR)
Foreign manufacturers must appoint an AIR to liaise with the CDSCO for regulatory compliance and submission management.
Submitting your Technical Documentation
Submission of a Device Master File (DMF) detailing design, materials, safety data, and intended use, along with a Plant Master File (PMF) outlining the manufacturing processes and quality systems compliant with ISO 13485:2016, is mandatory.
Preparing for regulatory review & approval
Class A and B devices are reviewed by Notified Bodies, while Class C and D undergo thorough CDSCO evaluations, including clinical assessments, conformity checks, and inspections. Expedited approvals may be possible using Free Sale Certificates from IMDRF/GHTF member countries.
Implementing Clinical Investigations
Clinical Investigations are required for high-risk devices without existing international approvals or those significantly modified in design, intended use, or target population, to validate safety and effectiveness.
Clinical trials in India are subject to ethical review by an Institutional Review Board (IRB), and approvals must be obtained from the Drugs Controller General of India (DCGI) before trials commence.
Achieving market authorisation
CDSCO issues a Manufacturing License (Form MD-5) for locally produced devices or an Import License (Form MD-15) for imported devices upon successful review.
What post-market requirements should be considered for the Indian market?
Post-market surveillance is an ongoing regulatory requirement in India, designed to ensure medical devices and IVDs remain safe and effective throughout their lifecycle. This real-world monitoring complements pre-market testing, uncovering insights that help manage risks and improve patient outcomes.
India’s evolving approach to post-market surveillance emphasises proactive risk management, improved data utilisation, and greater patient involvement, ensuring high safety standards in the rapidly advancing healthcare landscape.
Key components of India’s post-market surveillance include:
Adverse Event reporting
Manufacturers and importers must promptly report adverse events to the CDSCO. For Class C and D devices, Periodic Safety Update Reports (PSURs) are mandatory to evaluate ongoing risks and benefits.
Field Safety Corrective Actions (FSCAs)
Manufacturers must swiftly implement corrective measures, such as labelling updates or recalls, coordinating effectively with healthcare providers and regulators to maintain patient safety.
Post-market studies
CDSCO may require surveillance studies for high-risk devices or those with limited pre-market data, focusing on real-world performance, safety, and durability.
Advanced analytics
To overcome challenges like fragmented data and inconsistent reporting, leveraging predictive analytics and data-driven insights is crucial. This proactive approach helps anticipate device issues and enables timely risk mitigation.
Stakeholder collaboration
Enhancing collaboration among manufacturers, healthcare professionals, regulatory bodies, and patient advocacy groups fosters greater transparency, quicker responses to safety concerns, and improved compliance.
Patient engagement
Patient feedback on usability, performance, and unintended effects enriches surveillance, providing valuable real-world data to complement clinical evaluations.
What are the challenges unique to the Indian regulatory landscape?
Despite improvements in safety, efficacy, and quality, manufacturers still face several ongoing challenges within the Indian regulatory landscape:
Frequent regulatory updates
Regular policy changes require continuous monitoring, which can strain the resources of small and medium-sized enterprises (SMEs). Misclassification of devices further complicates approval processes, potentially delaying market entry.
International harmonization issues
While India aligns broadly with global standards like those of the IMDRF, differences persist, complicating simultaneous international market entries. Compliance with Good Manufacturing Practices (GMP) and maintaining robust Quality Management Systems (QMS) remain resource-intensive, particularly for smaller companies.
Infrastructure constraints
Limited accredited testing laboratories, clinical research facilities, and specialised supply chains can often delay product validation, regulatory approvals, and impact product availability and costs.
Pricing & reimbursement
India’s price-sensitive market demands innovative solutions for an affordable cost. Ambiguous reimbursement policies further complicate investment decisions and affect market confidence.
How to overcome the regulatory challenges in India
Be proactively engaged
Manufacturers should proactively engage with the CDSCO, utilising regulatory intelligence and digital compliance tools. More broadly, joining organisations like the Association of Indian Medical Device Industry (AIMED), can help businesses navigate regulatory challenges and advocate for policy reforms in the medical device sector.
Embrace collaboration
Strengthening infrastructure through public-private partnerships and increasing the number of accredited testing facilities can significantly streamline approvals.
Building industry-wide regulatory capacity through training programs and academia-industry collaboration is also crucial to address skill gaps, particularly among SMEs.
Utilise digital tools
Integrating artificial intelligence and data analytics into your compliance processes can significantly enhance efficiency and surveillance capabilities.
Partner with experts
Partnering with regulatory experts with thorough knowledge of the Indian regulatory landscape can help manufacturers overcome ambiguity and navigate the path to market access, saving time and money that can be allocated more constructively elsewhere.
In conclusion…
India’s regulatory framework for medical devices and IVDs continues to evolve, encompassing a robust, risk-based system that aligns with global best practices while addressing unique local requirements. Enhancements in regulatory transparency and harmonization, improved post-market surveillance facilitation, and expanded testing infrastructure will be crucial for the sustained growth of the Indian medical devices market. By addressing these factors, India will be well-positioned to become a leading global hub for medical device innovation and regulation.
For manufacturers, India’s regulatory environment can be challenging – but by maintaining regular engagement with the CDSCO, investing in compliance tools to assist with documentation maintenance, and collaborating with regulatory experts, India’s regulatory landscape can be navigated effectively.
Are you looking to register your medical device in India? Our Regulatory team is ready to help – simply get in touch to start the conversation.
Our fast-growing markets series continues…
Stay tuned for part four of our fast-growing markets series, which will explore the regulatory landscape in South Africa.