
It’s undeniable that adherence to EU MDR requirements remains a challenge for everyone involved. And with the complexity of the requirements increasing, adjusting to comply with the regulation comes at a cost.
But rather than seeing compliance with EU MDR as an unrecoverable additional expense with no benefit, adapting to the massive changes should instead be used as an opportunity to proactively evaluate existing processes. It’s a chance to find ways to improve the efficiency, simplicity, and connectedness of processes.
Simple, structured, and properly connected processes require less resources during maintenance, whilst ensuring the quality and safety of devices – increasing the satisfaction of users and improving the health of patients.
When it comes to EU MDR, the most important connections between different areas are arguably the interfaces between Risk Management, Clinical, and Postmarket Surveillance. In our topical article below, our Head of Regulatory and Clinical Corinne Larke outlines how to keep Risk, Clinical, and PMS connected under the EU MDR.
The benefits of connecting Risk, Clinical, and PMS under EU MDR
Whilst it may be tempting to put off the EU MDR work to avoid costs in the short term, waiting for audit findings to force change can be far more expensive in the long run. Being told from the outside what needs to change, retroactively, not only involves dedicating resources in a rush, but also having a solution dictated to your organisation from external sources that may not be the best fit. It can lead to unnecessary complexity that doesn’t add value and is difficult to maintain.
If executed sustainably, making the necessary changes can help your organisation be more efficient with resources, which ultimately leads to the manufacture of better medical devices. One important aspect is to ensure that the interfaces between processes are functioning. This enables the flow of data and avoids data silos that create duplication of work, under-utilise knowledge within an organisation, and make it more costly to arrive at the same or lesser result.
When used correctly, the connections between Risk Management, Clinical, and PMS lead to safer devices and more efficient use of existing data as it travels from one process to another. To enable this flow of data, connections must function appropriately, and be simple enough for easy maintenance. When creating the connections, it’s important to consider solutions that really function for your individual organisation.
To maintain a functioning exchange of data, it’s crucial to understand how the requirements between Risk, Clinical and PMS are connected.
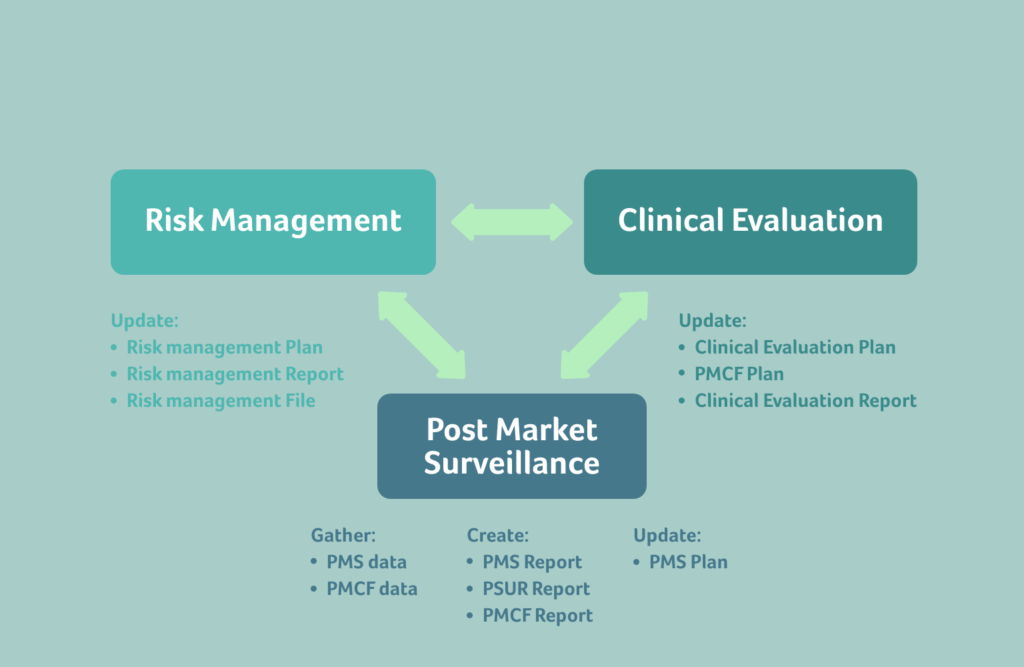
From PMS to Risk: Focus on managing knowledge and recognising trends
When gathering PMS data and creating PMS or Periodic Safety Update (PSUR) reports, it’s important to focus on managing and actively using knowledge in other areas of your organisation, to improve current and future device iterations.
The PMS data must be able to easily enter the Risk Management process, and continuously keep the benefit-risk determination of the device updated. This will ensure that your risk management files are living, meaningful documents. Postmarket data that enters the Risk Management system enables proactive corrections instead of costly, reactive field actions, recalls, or corrective actions. And it ensures that future devices avoid issues found through current PMS data. Collecting PMS data simply because it’s a requirement under the EU MDR, without utilising it any further in other areas, is a costly exercise without benefit.
PMS data for one device must also impact PMS activities for other similar devices, to help the similar devices currently on the market to avoid the same issues. Following this requirement helps to proactively look at devices and pre-empt potential issues.
One of the ways for PMS data to connect to Risk is via the trending PMS data. It’s important to develop and utilise a meaningful statistic methodology to detect statistically significant increases or trends of postmarket incidents, and the approach must be documented in your PMS plan. Suitable indicators and threshold values will show when a trend is impacting the benefit-risk determination of the device, which in turn will impact the risk control measures for the device.
Having a proactive and systematic approach, as well as effective processes for data collection and analysis, will ensure that all collected data is utilised for fulfilling the MDR requirements for Risk and PMS. More importantly, this kind of approach will allow early intervention regarding any issues that may impact the benefit-risk profile – avoiding the costs associated with reactive recalls or field actions.
PMS data is also intended to monitor residual risks of the medical device that remain when it’s placed on the market, after all other risks have been reduced as far as possible. PMS data is used to ensure the risk levels associated with a device remain updated based on real world events, and that key risk indicator trends are tracked for risks that adversely affect the benefit-risk ratio of the device. It is used for failure modes and trends benchmarking, and for early recognition of emergent risks.
From Risk to PMS: Ensure thorough postmarket risk management
Risk influences how PMS activities are performed, and within EU MDR, postmarket risk management is generally more emphasised.
The risk class of the device determines the type of PMS activities that must be planned for the device, i.e., the PMS system must be risk based.
Investigations of serious postmarket incidents must include a risk assessment of the incident, which will determine further actions, and the trending of incidents is also risk based.
From PMS to Clinical: Continuously update your Clinical Evaluations with PMCF and PMS data
Clinical evaluations must be continuously updated with clinical data obtained from PMCF activities and through the PMS plan.
Along with PMCF data, PMS data must also feed into clinical evaluation reports throughout the life cycle of the device.
From Clinical to Risk: Consider Clinical safety in your CEP, CER, and PMCF
In EU MDR, there isn’t only a flow of data from PMS data to risk, but also from Clinical data into risk management.
Your Clinical Evaluation Plan must contain methods used to examine clinical safety and it should be transparent regarding how residual risks are determined. The acceptability of the benefit-risk ratio must be based on the state of the art for that indication or intended use of the device.
Undesirable side effects found through Clinical investigations must be analysed, using the methods from the plan, to determine if they constitute acceptable risks when weighed against the benefits of the device.
The Postmarket Clinical Follow up (PMCF) process must continuously, throughout the lifetime of the device, confirm that the device risks are still acceptable. It needs to detect emerging risks, and this data should flow into the risk management process. The PMCF plan must lay out methods for how emergent risks will be identified and analysed. Furthermore, the PMCF should ensure that the benefit-risk ratio of the device remains acceptable.
The PMCF plan must be connected to the Clinical Evaluation Report (CER), and the CER must connect to the risk management. In turn, the conclusions of the PMCF evaluation report should also feed into risk management.
Your Clinical Investigation report must consider and assess the risks versus clinical benefits of your medical device.
From Risk to Clinical: Reflect the device benefit-risk ratio in your clinical investigation plan
As with PMS, clinical activities are also influenced by the risk of the device.
The depth of the Clinical Evaluation Report (CER) should be in proportion to the risk of the device.
The clinical investigation plan needs to reflect aspects related to the benefit-risk ratio of the device and consider known or foreseeable risks.
During product development, the risk will inform the type of clinical data required, as well as whether a trial or study are needed.
In conclusion…
Understanding and managing the data flow within your organisation is the first step towards maintaining functioning connections between Risk Management, Clinical and PMS. If executed carefully and correctly, and in a way that is right for your individual organisation, the data created can be of value for current and future devices, as well as for users and patients alike.
Ensuring that your system is clear and simple, makes its maintenance easy throughout the lifecycle of your devices, and enables warning signs from one area to trigger the right actions in other areas – ultimately creating a value-adding, sustainable QMS.
Should you have a challenge related to Risk, Clinical or PMS – or adherence to the EU MDR more generally, our Regulatory and Clinical teams are ready and happy to help. Simply get in touch to start the conversation.

